El objetivo de esta tesis es medir si es viable el tratamiento genético que
propone Sarepta Therapeutics, este será un análisis largo y
denso. El fin es ser cautelosos con los riesgos y posibles upsides, ya que
emitir mal juicio en cualquiera de estos aspectos podría ser desastroso:
por una parte perdemos una gran parte de retorno y la otra una posición
muy grande, distorsionada por el retorno/riesgo, provocará perdidas
que se pudieron haber evitado. Esas son las razones de este análisis, un tanto
simplista, pero espero que sea lo más denso y sopesado en mis posibilidades.
Está será la estructura, por si interesa una parte más que otra y solo
quieres saltar a leerla:
Con la estructura aclarada, espero que el análisis sea interesante y presente
una guía de mi toma de decisiones y a futuro poder hacer autocrítica y mejorar.
1. Introducción y Perfil de la Compañía
Sarepta es una compañía de biotecnología con el objetivo de desarrollar
medicina genética de precisión. Comenzó como AntiVirals el 1980, cuando
hicieron su IPO cambiaron su nombre a AVI BioPharma y el 2012 a Sarepta Therapeutics.
En sus inicios era una empresa que se dedicaba a la investigación y desarrollo
de anti-virales, el 2000 comenzó el trabajo de su tecnología antisense
NeuGene siendo el pivote a su plataforma de tecnología genética. El año 2003. anunciaron intentos de desarrollo de tratmientos para el SARS y el
virus del Nilo Occidental. El 2006 comenzó a proponer estudios de
concepto para un medicamento RNA experimental en DMD, llevando
su atención hacia enfermedades neuromusculares. Durante
el 2009 estando a punto de quebrar recibieron un contrato de $11.5m del
Departamento de Defensa de Estados Unidos.
El 2012 se renombró como Sarepta Therapeutics ($SRPT), reflejando
su cambio estratégico hacia tratamientos genéticos de precisión para
trastornos neuromusculares raros, especialmente DMD. También el
cambio pudo haber sido propulsado por sus desarrollos en la plataforma
PMO usados para el tratamiento de influenza, dengue, SARS, hepatitis C.
Para 2016 con la aprobación del PMO Exondys 51 Sarepta continuó
expandiéndose y su desarrollo de terapia genética para Duchenne con más
PMOs skippers de exones y Elevidys.
Su cambio estratégico se centró en tratamientos genéticos para trastornos raros, especialmente la Distrofia Muscular de Duchenne (DMD).
2. La Enfermedad: Duchenne Muscular Dystrophy (DMD)
La enfermedad de Distrofia Muscular Duchenne se trata sobre la incapacidad
de generar distrofina de forma natural, provocando la muerte de los
músculos que más la necesitan desde el momento de concepción del niño. La esperanza de vida ha mejorado durante las décadas, con algunos hombres
llegando a vivir 40 años gracias a la evolución de sus tratamientos y la
medicina: cuidados y mediciones meticulosas de los pacientes junto a
tratamientos de corticoides permiten que la esperanza de vida haya mejorado
de 10-15 años hasta 30-40 años, aunque es una mejora no es suficiente. La
calidad de vida se ve altamente deteriorada por esta condición, no afecta solo
habilidades motrices, sino que también a capacidades respiratorias y
cognitivas. Esto es la principal misión del Elevidys de Sarepta: en el mejor
de los casos detener el progreso del deterioro en niños.
El DMD a estado presente en la humanidad desde siglos atrás, se
sospecha de diagnósticos desde Egipto antiguo en pinturas de tumbas datadas
a 2800-2500 A.C hasta una pintura que el mismísimo Duchenne diagnosticó
con la condición: Transfiguración de Rafael.
Propiedades Genéticas del DMD
Esta afección es heredada por el cromosoma X, habitualmente de una madre
portadora, las mujeres en esta condición no suelen ser fenotipo, pero existen
casos. Siendo más preciso la locus del genoma es en Xp21, desde Xp211 hasta Xp213 compartiendo vecindad con otras mutaciones genéticas
como el BMD y demás – se tratará similitudes más adelante. Haciendo que
el locus de la distrofina sea de ~3mb, cerca de un 3% del cromosoma X.
Esta es una de las hipótesis de su alta recurrencia en mutaciones: al ser
un gen tan grande es más posible un daño en el genoma, como pueden
ser deleciones, duplicaciones y mutaciones puntuales (~70%, 10-15% y
~15% respectivamente en los casos).
Estas mutaciones en el genoma encargado de la de la fase de la proteína
causa que sea imposible la producción de una distrofina funcional, que
es necesaria para musculo esquelético y suave.
Función de la Distrofina
La distrofina se encuentra en el sarcolema y miotubos maduros, menos
en gente con Duchenne. Estas personas tienen una resistencia reducida
a shock osmótico, debido a la ausencia de distrofina en los miotubos
indicando una “fragilidad” (pp. 131 y 173) de los sarcolemas de células
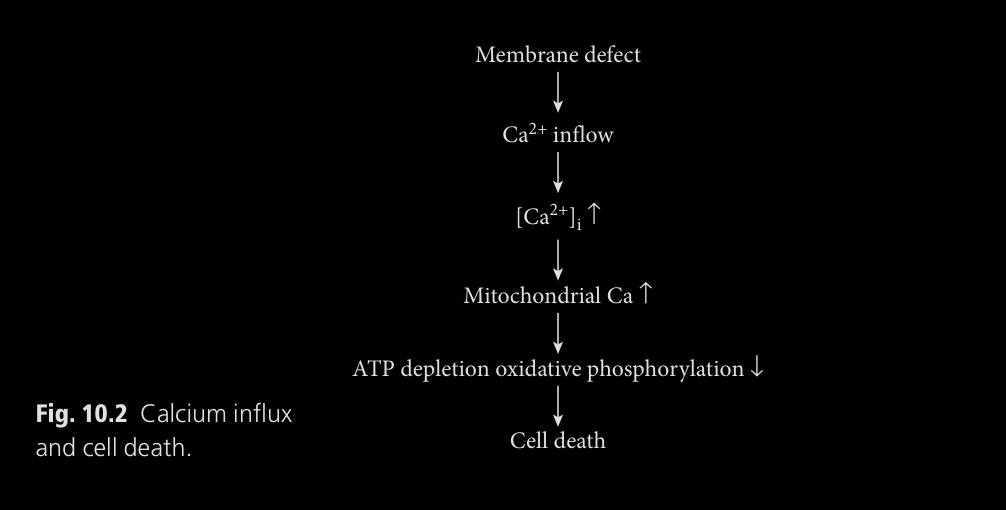
musculares con DMD. Una célula sana es capaz de soportar estos shocks osmóticos gracias
a la distrofina, que son causados por el alargamiento del músculo
bajo carga – contracciones normales. La membrana sufre micro-roturas
que permiten la fuga de CK-MM y la entrada de calcio intracelular.
Otro efecto es la degradación de la duplicación de células satélite
(miogenicas) necesarias para la regeneración de las roturas de células
musculares mediante las células satélite.
La entrada de calcio en en exceso activa enzimas que degradan proteínas
(proteasas) y sobrecargan las mitocondrias agotado su ATP. El daño acumulado lleva a necrosis en las células musculares, que con el tiempo
se vuelve imposible con la degradación de las células satélites. Finalmente,
el tejido perdido es reemplazado por fibrosis y tejidos adiposos, lo que
lleva a la debilidad típica de DMD y su visualización al estar avanzada.
Tejidos Distintos a Músculo Esquelético Afectados
Los tejidos afectados, en parte, son el musculo cardiaco, liso, el cerebro, y
posiblemente la retina.
Músculo Liso
Ileus paralitico, volvulus y dilatación gástrica han sido reportados y suelen
ser eventos terminales. Constipación severa y fisuras anales han sido
reportados en adultos con DMD. Ocasionalmente hay presencia de
diabetes tipo 2 (por la no absorción de glucosa al no existir suficiente
musculo) y parálisis de vejiga. Estudios animales demostraron la
presencia de distrofina en músculos endoteliales lisos junto a deficiencia en
adhesividad de plaqueta y agregación inducida por ristocetina, que
explicarían (son solo hipótesis) la común perdida de sangre severa por
parte de niños con DMD en operaciones mayores.
Músculo Cardíaco
Desde temprana edad hay taquicardia sinusal persistente. Después de
perder independencia ambulatoria se presenta cardiomiopatia, aunque solo
un 15% de los pacientes desarrollan insuficiencia cardiaca progresiva, todos
muestran signos cardiacos en etapas avanzadas.
Se presenta una fibrosis miocardico en el posterobasal del ventrículo izquierdo, aunque hay evidencias de presencia en el derecho. El proceso
de evolución de tejidos del corazón es similar al esquelético, reemplazo de
musculo por tejido conectivo y grasa.
Sistema Vascular
En niños con DMD encontraron que su circulación se encuentra sobre o
debajo del rango 95% para niños sanos. Más reciente es el descubrimiento
de reducido flujo sanguíneo intramuscular en ratones y niños con DMD.
Es posible por la ausencia de Óxido Nítrico Sintasa (NOS en inglés), y,
por ende, Óxido Nítrico en el musculo esquelético sin distrofina. Una de
las proteinas relacionadas con la distrofina es el NOS neuronal (nNOS).
Creo que es mejor que el libro explique esta parte:
"This molecule, physiologically expressed at the periphery of each
muscle fibre, is lost from the sarcolemma when dystrophin is also absent.
One physiological role of nNOS in skeletal muscle is the production of
NO that mediates the inhibition of sympathetic vasoconstriction in contracting muscle. This ability is defective in the mutant mice that lack
the gene encoding nNOS, and also in the mdx mouse in which nNOS is
also secondarily, but severely, deficient. Recently, a similar defect was
confirmed in children with DMD. These observations also suggest
another mechanism that might contribute to abnormal smooth muscle
function in DMD, and eventually to muscle fibre necrosis."
Sistema Nervioso Central
La severidad y frecuencia de retraso mental crece con una perdida
progresiva de isoformas distales funcionales. También, ADHD se
presenta con mutaciones que afecten la isoforma cerebral de las
isoformas de distrofinas Dp140 y Dp71. Al mismo tiempo se nota
Autismo, déficit de memoria y problemas de comportamiento.
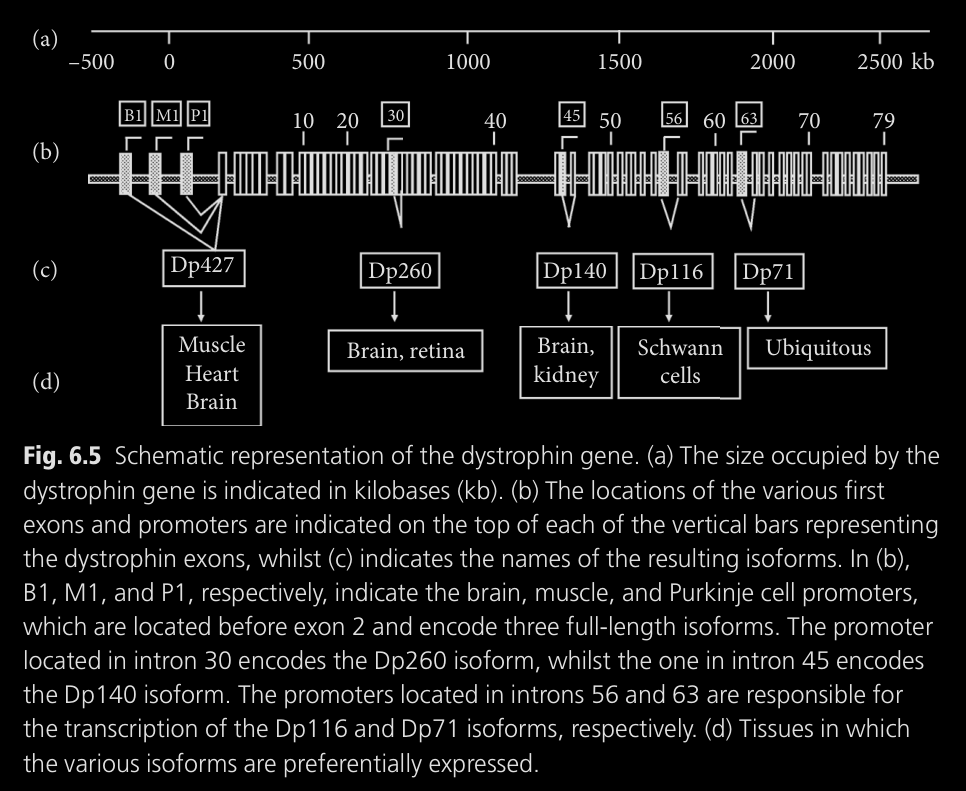
Las deleciones de exones en el ARN de producción de la distrofina
provoca distrofinas mal funcionales que, por consecuencia,
producen isoformas cerebrales de la distrofina que también son
inútiles. Como el gen de distrofina es tan largo, distintas deleciones
en sectores del gen provocaran isoformas falladas con objetivos
distintos en distintos tejidos:
Deleciones de los exones 1-44 afectan las isoformas del cuerpo completo
y a la Dp260. En este caso las isoformas cerebrales más cortas siguen
funcionando (Dp140 y Dp71).
La gráfica del libro lo explica mucho mejor:
Sistema Esquelético
En personas con DMD también hay problemas en el esqueleto, pero es
debido a el no uso de los músculos. Al no existir tensión de las fibras
musculares el hueso de se deforma y pierde densidad.
Otros
El oído no se ve afectado. Hiperplasia del timo se presenta por varios
autores, causas desconocidas. Pubertad suele ser atrasada, empeorado por
el uso de corticoides. Hiperestrogenemia puede darse, y obesidad es
frecuente, de nuevo más común en niños tratados con corticoides.
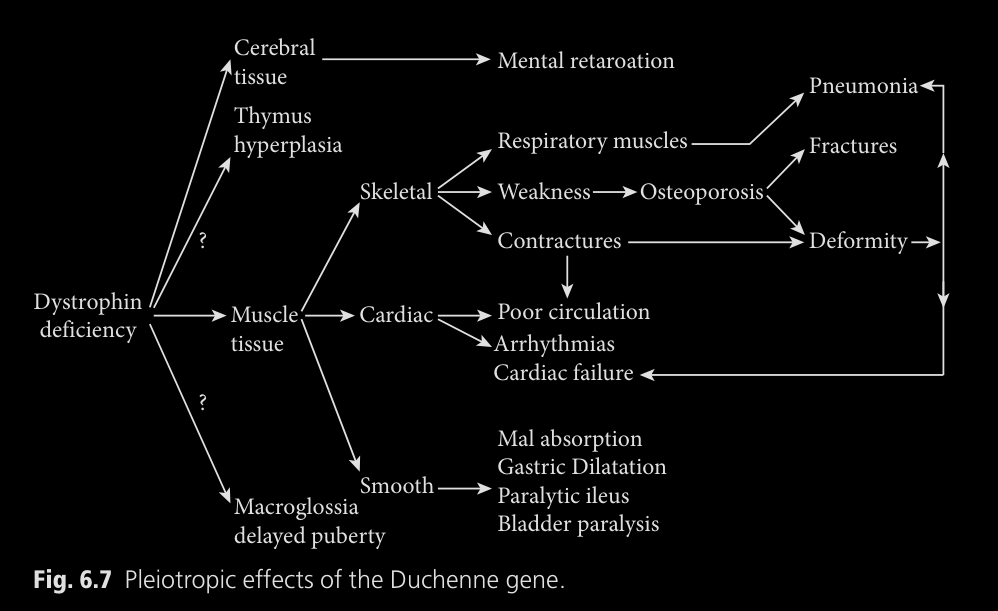
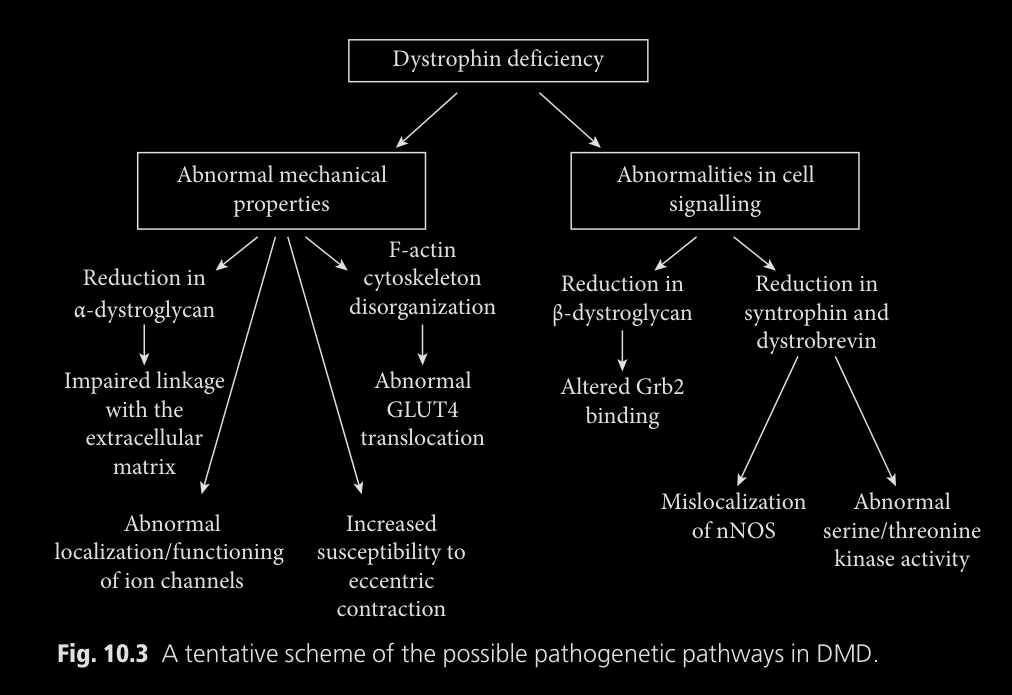
Lo descubierto seria representado en este diagrama:
Los efectos de la deficiencia de distrofina podrian ser representados en este
diagrama, si es necesario ulgún tipo de aclaración lo mejor es leer el libro
del que estudié la enfermedad: Duchenne Muscular Dystrophy (Oxford Monographs on Medical Genetics)
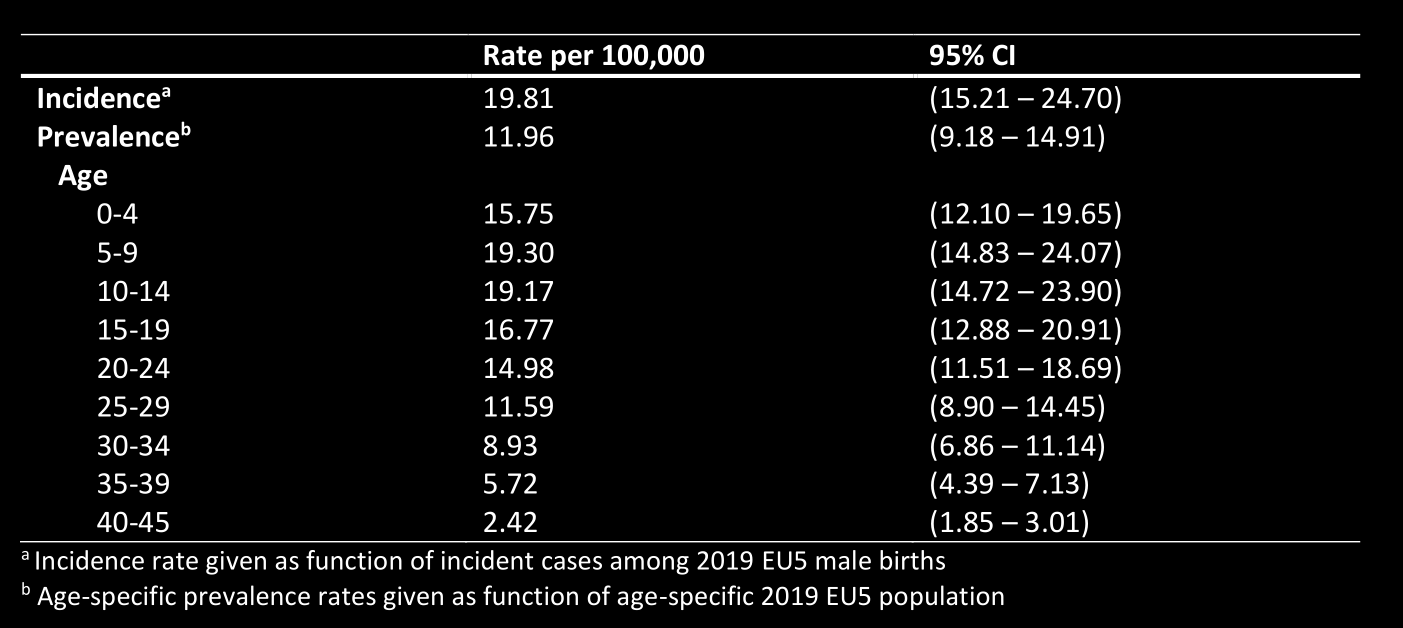
Con lo que es posible ver dos cosas: 1. es difícil superar los 20 años y 2.
los casos son descubiertos entre los 5-9 años. Tomando en cuenta este
como un indicador de nuevos casos quedaría en un ratio de ~1:5181 niños.
Cerca del número que se usa como estimaciones.
Estados Unidos presenta una tasa parecida si es que no igual. Por lo que es
posible concluir que es una tasa de 1 cada 5000 niños que padecen
Duchenne.
3. PMOs de Sarepta
EXONDYS 51 (eteplirsen)
Exondys 51 fue aprobado por la FDA el septiembre del 2016, fue
controversial dado a resultados y metodologías dudosas por parte de
Sarepta.
Mecanismo de Acción
Antes de la producción de la proteína existe el proceso de lectura y copia
del gen de distrofina del ADN, que es el pre-ARNm. El spliceosoma
lee secciones especificas en cada exón, une secuencialmente los exones,
elimina entrones creando ARNm maduro. Con la falta de algún tipo de
exón la traducción se aborta y no hay distrofina funcional.
El eteplirsen se encarga de entrar al núcleo de la celula muscular y se
hibridiza dentro del exón 51 del preARNm, bloqueando fisicamente el
acceso del spliceosoma a esa región, reemplazando el exón 51 y haciendo
la conexión del último disponible con el exón 52. Por eso es una estrategia de
skipping, se salta la lectura del exón faltante, permitiendo la lectura
y traducción de un gen incompleto de distrofina.
Como vimos anteriormente, los efectos de la falta de distrofina no es
solo musculo-esquelético e inclusive depende de la falta de exones la
no existencia de distintos isoformas de la proteína necesarios para
distintas células de distintos órganos. Los isoformas que se ven afectados
son el Dp427, Dp260 y el Dp140. En los dos primeros casos se ve una
versión más corta de sus isoformas (Dp427-Δ51 y Dp260-Δ51,
respectivamente), mientras que el Dp140 no es producido, ya que su
codón de inicio está dentro del exón 51. Dp140 no se produce, usado
en tejidos del cerebro, riñón y más).
Enzayos Clínicos
En los primeros ensayos se demostró que es tolerable, ya que es de baja
toxicidad, no hay respuesta inmune, por ende alta penetración al
músculo, no penetra barrera cerebral.
Hablando de resultados, las cosas cambian un poco. Hay que tener
en mente que esta enfermedad es muy individual, muy distinta entre
cada persona, también la evolución – distinta en gravedad y efectos
en cada paciente. Lo que es más o menos constante es que el deterioro
es notorio mientras más tiempo pase. El libro también aclara que el
quedar incapacitado y, por tanto, el uso de silla de ruedas, es un predictivo
de muerte en unos años más para el niño, que sin corticoides y otros
cuidados no alcanzaban la adultez. Es importante tener esto en mente.
NCT02255552: Study PROMOVI
Primary | Time Frame: Week 96 | Unit: Meters
1. Change from Baseline in 6 Minute Walk Test (6MWT)
Standardized 25-meter course; distance recorded over 6 minutes.
Group
Mean (SD) Change
Eteplirsen 30 mg/kg
-117.91 (128.488)
Untreated Control Group
-133.56 (129.333)
Secondary | Time Frame: Week 96
2. hange From Baseline in Dystrophin Protein Levels Determined by Western Blot at Week 96
Change from baseline in dystrophin protein levels (in muscle biopsy samples) were determined by Western blot. For each time point, 2 blocks of tissues were analyzed by Western blot, each with 2 replicates of gels to determine the dystrophin level as compared to a healthy individual (Percent Normal). The block average value from 2 replicate gels was computed. The overall average was calculated as the mean of the block average values. The overall average values were used for all analyses. In case only 1 gel was available for a block, then that value was used as the block average value.
Group
Mean (Standard Deviation) | Unit of Measure: Percent Normal Dystrophin Protein Level
Eteplirsen 30 mg/kg
0.516(0.7236)
Untreated Control Group
-
Secondary | Time Frame: Week 96
3. Ability to Rise Independently (NSAA)
NSAA is a clinician-administered scale that rates participant performance on 17-items and included assessments of abilities such as 10-meter walk/run, rising from a sit to stand, standing on 1 leg, climbing a box step, descending a box step, rising from lying to sitting, rising from the floor, lifting the head, standing on heels, and jumping. For all activities, participants were graded as follows: 0 = unable to achieve goal independently; 1 = modified method but achieves goal independent of physical assistance from another and 2 = normal, no obvious modification of activity. Number of participants having ability to rise independently from the floor indicated by a NSAA Rise from floor sub score greater than 0 (unable to achieve goal independently) was reported.
Group
Participants (n / %)
Eteplirsen 30 mg/kg
33 | 54.1%
Untreated Control Group
33 | 3.3%
Secondary | Time Frame: Week 96
4. Number of Participants Who Lost Ambulation (LOA) by Week 96
-
Group
Participants (n / %)
Eteplirsen 30 mg/kg
12 | 17.9%
Untreated Control Group
1 | 5.0%
Secondary | Time Frame: Week 96
6. Change From Baseline in North Star Ambulatory Assessment (NSAA) Total Scores at Week 96
-
Group
Mean (Standard Deviation) | Unit of Measure: Unit on scale
Eteplirsen 30 mg/kg
-7.23 (5.173)
Untreated Control Group
-8.44 (9.812)
Secondary | Time Frame: Week 96
7. Change From Baseline in Dystrophin Intensity Levels Determined by Immunohistochemistry (IHC) at Week 96
-
Group
Mean (Standard Deviation) | Unit of Measure: Percent dystrophin positive fibers
Eteplirsen 30 mg/kg
0.030 (0.0360)
NCT01540409 – Efficacy, Safety, and Tolerability Rollover Study of in Subjects With Duchenne Muscular Dystrophy
Primary | Time Frame: Week 240 | Unit: Meters
1. Change from Baseline in 6 Minute Walk Test (6MWT)
Standardized 25-meter course; distance recorded over 6 minutes.
Group
Mean (SD) Change
Eteplirsen 30 mg/kg
-199.0(113.25)
Eteplirsen 50 mg/kg
-258.0(175.65)
Primary | Time Frame: Week 48
2. Change From Baseline in the Percentage of Dystrophin Positive Fibers (PDPF) at Week 48
Change from baseline in dystrophin protein levels (in muscle biopsy samples) were determined by Western blot. For each time point, 2 blocks of tissues were analyzed by Western blot, each with 2 replicates of gels to determine the dystrophin level as compared to a healthy individual (Percent Normal). The block average value from 2 replicate gels was computed. The overall average was calculated as the mean of the block average values. The overall average values were used for all analyses. In case only 1 gel was available for a block, then that value was used as the block average value.
Group
Mean (Standard Deviation) | Unit of Measure: Percent Normal Dystrophin Protein Level
Placebo to Eteplirsen
37.70(12.602)
Eteplirsen 30 mg/kg
51.69(7.089)
Eteplirsen 50 mg/kg
42.93(13.433)
NCT06606340 – A Long-term Observational Study Evaluating Eteplirsen, Golodirsen, or Casimersen in Routine Clinical Practice (EVOLVE)
Iff J, Desguerre I, Liu Y, et al. Association Between Exon-Skipping Therapy With Eteplirsen and Cardiac Outcomes in Duchenne Muscular Dystrophy. Presented at: 2025 MDA Clinical & Scientific Conference; March 16-19. Dallas, TX. Abstract P79
Abstract
BackgroundDuchenne muscular dystrophy (DMD) leads to dilated cardiomyopathy and heart failure during teenage years or young adulthood. Eteplirsen promotes dystrophin production through skipping of exon 51 of the DMD gene.ObjectiveThis analysis compared LVEF decline between eteplirsen-treated and control patients with exon 51 skip-amenable DMD.MethodsEteplirsen-treated patients from clinical trials were matched with control patients from natural history studies in a propensity score analysis. Risk of reaching LVEF thresholds (50%, 55%, and 60%) was evaluated using Cox proportional hazard models. Annual rate of LVEF decline was characterised using linear mixed effects models.ResultsAmong 141 eteplirsen-treated and 103 control patients available for matching, the analysis included 122 eteplirsen-treated patients matched with 122 control patients (64 unique control patients). No eteplirsen-treated and 27 controls (22.1%) reached LVEF <50%; eteplirsen-treated patients had a lower risk of reaching <55% and <60% thresholds versus controls (hazard ratio = 0.22; 95% CI = 0.07-0.66; P < 0.01 and hazard ratio = 0.40; 95% CI = 0.22-0.76; P < 0.01, respectively). Annual rate of LVEF decline for eteplirsen-treated and controls was -0.66% (95% CI = -0.96 to -0.36, P < 0.01) and -1.38% (95% CI = -1.60 to -1.16; P < 0.01), respectively. Results were consistent with a sensitivity analysis matching each eteplirsen-treated patient once with a unique control patient and with several tests for potential bias.ConclusionsIn this retrospective study, eteplirsen-treated patients were observed to have a significantly lower risk of reaching LVEF thresholds indicative of cardiac function decline and attenuation of LVEF decline compared with matched controls.
Mathews K, Grabich S, Dharmarajan S, et al. Comparative Analysis of Loss of Ambulation in Eteplirsen-Treated Patients With DMD in the EVOLVE Study and Propensity Score–Weighted External Controls. Presented at: 2025 MDA Clinical & Scientific Conference; March 16-19. Dallas, TX. Abstract P103.
Background: The Phase 4 EVOLVE study (NCT06606340) evaluates phosphorodiamidate morpholino oligomers for males with Duchenne muscular dystrophy (DMD) in routine clinical practice. The average age at loss of ambulation (LOA) in patients with exon 51 skip-amenable DMD is ~12 years.
Objectives: To examine the effect of eteplirsen on LOA by comparing patients from EVOLVE to propensity score–weighted external controls (ECs) receiving standard-of-care corticosteroids.
Methods: The study population included male, exon 51 skip-amenable DMD patients ambulant at baseline. Key baseline confounders were balanced in the eteplirsen-treated and EC groups using inverse probability treatment weighting (IPTW), including age (4–7 or 8+ years), corticosteroid use duration (≥1 or <1 year), 10-meter walk/run (10MWR) time velocity, and rise from floor velocity. LOA was defined as the earliest date of patient-/caregiver-reported continuous wheelchair use, verified by attending physician, North Star Ambulatory Assessment walk score of 0 or 10MWR score of ≥30 (if available), or inability to perform 10MWR, and not a temporary event. Results: 33 eteplirsen-treated patients and 75 ECs, derived from 5 EC groups (CINRG-DNHS, FOR-DMD, PRO-DMD-01, Leuven NMRC, Italian DMD Telethon) met inclusion criteria. After IPTW, LOA occurred in 21/33 eteplirsen-treated patients (13.5/100 patient-years [PY]; 4.7-year mean follow-up) and 19/33 weighted ECs (20.8/100 PY; 2.9-year mean follow-up). For those who experienced LOA through the interim analysis, median (95% CI) age at LOA by Kaplan-Meier analysis was 15.3 (11.4–15.8) years in eteplirsen-treated patients vs 11.3 (8.0–12.8) years in ECs. Cox proportional hazard regression analysis suggested eteplirsen reduced LOA risk by 62% (95% CI, 20–82; P=0.011) across the lifespan. We also analyzed patient differences at age 15 for an understanding of eteplirsen treatment impact at a critical point of disease progression. Nonparametric analysis estimated that the probability (95% CI) of remaining ambulant was 0.50 (0.32–0.69) and 0.14 (–0.06 to 0.34) at age 15 for eteplirsen-treated patients and ECs, respectively. Treatment with eteplirsen provided a 36% (95% CI, 9–63) higher likelihood of remaining ambulant by age 15. Conclusions: This interim analysis shows a clinically and statistically significant delay in LOA in eteplirsen-treated patients vs ECs using advanced methods to reduce confounding in a real-world setting.
Conclusiones
Es comprensible la decisión de la FDA de no aceptar el tratamiento por
parecer inservible, pero como demuestran los resultados a largo plazo,
hubiese sido un grave error, no solo porque no habría sido posible darle
el necesario tratamiento de Exondys a personas que lo necesitaban, sino
que hubieran provocado la bancarrota de Sarepta y la posibilidad de la
no producción del resto de PMOs por miedo a no tener resultados a corto
plazo. La Dra. Janet Woodcock acertó con su intuición y actuar, junto a
la organización de las familias que lograron la aprobación acelerada.
Ahora, hablando de números. Sí, baja producción de niveles de distrofina,
menos del 1%, pero aparentemente mucho mejor que el casi o 0% de la
ausencia del tratamiento. Como se ve a largo plazo con EVOLVE se
redujo en 62% el riesgo de pérdida de marcha, que con el libro, sabemos
que es antecesor a problemas en pulmones (incluso infecciones) y después
el corazón, culminando en muerte. La pérdida de ambulación pasa de 11.3 a
15.3 años. Se preserva la función cardiaca ya que reduce significativamente
el declive de la eyección del ventrículo izquierdo (que el libro dice ser
predecesor del derecho). Además, mantenimiento de función de los
miembros superiores en el 78% de los casos.
El tratamiento funciona y podría hacer un análisis del resto de PMOs, pero
lo considero innecesario, ya que están aprobados y lo que nos concierne no es
el pasado, y quizá ni siquiera el presente, sino que el futuro. Por ahora,
me referiré a los PMOs como la caja de Sarepta y lo que lo mantiene a
flote con todas estas caídas en bolsa, exageradas en mi opinión.
4. Elevidys
Estados de Ciertos Estudios
ENVOL, a Phase 2, open-label trial evaluating the safety and expression
of delandistrogene moxeparvovec in patients with Duchenne muscular
dystrophy aged <4 years: Study design
ENVOL (Study 302) Temporarily Halted
SRP-9001-104 Temporarily Halted
SRP-9001-303 Temporarily Halted
Esto por muertes en no ambulatorios.
Elevidys o delandistrogene moxeparvovec es una transferencia génetica
por medio de un vector no replicante (rAAVrh74) con alto tropismo a
tejido muscular esquelético y al miocardio. La administración solo es
permitida ante ausencia de lecturas elevadas de anticuerpos anti-rAAVrh74,
mayor a 1:400. Lo que este virus hace es penetrar en los miocitos. El material genético no
se integra al ADN del paciente, sino que queda en el núcleo de la célula
muscular como un episoma estable.
Micro-distrofina
No es posible – hasta ahora – una distrofina completa, dado que el AAV no
es capaz de incorporar los ~3 megabases del ADN de la distrofina (recordar
que es el más grande en el código genético humano) y 14 kilobases del
ARNm. Los AAV tienen una capacidad máxima de ~4.7 kilobases.
La proteína resultante es una expresión más corta de la distrofina de 138 kDa
que conserva dominios críticos de la versión nativa de 427 kDa. Los
dominios son el de unión a la actina en el extremo amino-terminal y los dominios de unión al complejo de glicoproteínas asociadas a la distrofina (DAGC) en el extremo carboxílico-terminal. En español, esto significa que
se une a la actina F del citoesqueleto interno y unirse al DAGC manteniendo
los dos extremos y, como penetra musculo esquelético y corazón, permite
la conexión de la micro-distrofina en estos objetivos. Es decir, estabiliza
al Sarcolema y permite atrasar la necrosis de estos tejido.
Lamentablemente el vector no es capaz de penetrar el cerebro, por lo que
el sistema nervioso no podrá tener isoformas necesarios para las células
nerviosas.
5. Resultados de Clínicos de Elevidys
NCT03375164 – trial 101
Este fue un ensayo de fase inicial ½, abierto y no aleatorio. Cuatro niños de
entre 4 y 7 años.
Resultados a 2020, de pubmed.ncbi.nlm.nih.gov/32539076/
Bien tolerado y mínimos eventos adversos, expresión robusta y correcta
localización.
Results: Four patients were included (mean [SD] age at enrollment, 4.8 [1.0] years). All adverse events (n = 53) were considered mild (33 [62%]) or moderate (20 [38%]), and no serious adverse events occurred. Eighteen adverse events were considered treatment related, the most common of which was vomiting (9 of 18 events [50%]). Three patients had transiently elevated γ-glutamyltransferase, which resolved with corticosteroids. At 12 weeks, immunohistochemistry of gastrocnemius muscle biopsy specimens revealed robust transgene expression in all patients, with a mean of 81.2% of muscle fibers expressing micro-dystrophin with a mean intensity of 96% at the sarcolemma. Western blot showed a mean expression of 74.3% without fat or fibrosis adjustment and 95.8% with adjustment. All patients had confirmed vector transduction and showed functional improvement of NSAA scores and reduced creatine kinase levels (posttreatment vs baseline) that were maintained for 1 year.
Conclusions and relevance: This trial showed rAAVrh74.MHCK7.micro-dystrophin to be well tolerated and have minimal adverse events; the safe delivery of micro-dystrophin transgene; the robust expression and correct localization of micro-dystrophin protein; and improvements in creatine kinase levels and NSAA scores. These findings suggest that rAAVrh74.MHCK7.micro-dystrophin can provide functional improvement that is greater than that observed under standard of care.
NCT03769116 – trial 102
Ensayo clínico de fase 2, aleatorio, doble ciego y controlado por placebo.
41 pacientes de 4 a 7 años. Dos fases: una controlada por placebo de 48
semanas y otra fase de cruce (crossover) donde el grupo de placebo
recibe la terapia.
Expresión robusta de micro-distrofina en sarcolema a semana 12 del 23.82%
con p-value<0.0001. NSAA sin mejoría estadística a las 48 semanas, análisis
post-hoc sugirió desequilibrio entre subgrupos 6-7 años y 4-5 años, donde
los mas jóvenes monstruo mejoría estadísticamente significativa con P-Value= 0.0172 y NSAA 4.3(0.6). Mientras que el primer grupo P-Value= 0.5384 y NSAA -0.2(0.7).
Results: The 1-year safety profile of commercial process delandistrogene moxeparvovec in ENDEAVOR was consistent with safety data reported in other delandistrogene moxeparvovec trials (NCT03375164 and NCT03769116). Delandistrogene moxeparvovec micro-dystrophin expression was robust, with sarcolemmal localization at week 12; mean (SD) CFBL in western blot, 54.2% (42.6); p < 0.0001. At 1 year, patients demonstrated stabilized or improved North Star Ambulatory Assessment total scores; mean (SD) CFBL, +4.0 (3.5). Treatment versus a propensity score-weighted external natural history control demonstrated a statistically significant difference in least squares mean (standard error) CFBL in North Star Ambulatory Assessment, +3.2 (0.6) points; p < 0.0001.
Interpretation: Results confirm efficient transduction of muscle by delandistrogene moxeparvovec. One-year post-treatment, delandistrogene moxeparvovec was well tolerated, and demonstrated stabilized or improved motor function, suggesting a clinical benefit for patients with Duchenne muscular dystrophy. ANN NEUROL 2023;94:955-968.
NCT04626674 – ENDEAVOR, trial 103
Primary (Current) (Submitted: 2025-12-19)
Parte 1 (Cohortes 1 a 5): Cambio desde el inicio en la cantidad de expresión de distrofina de delandistrogene moxeparvovec a la semana 12, medido por Western Blot [Marco de tiempo: Inicio, Semana 12].
Parte 1 (Cohortes 6 a 8): Cantidad de expresión de distrofina de delandistrogene moxeparvovec a la semana 12, medido por Western Blot [Marco de tiempo: Semana 12].
Cohorte 8: Número de participantes con lesión hepática aguda (ALI) [Marco de tiempo: Inicio hasta la semana 72].
Secondary (Current) (Submitted: 2025-12-19)
Eliminación del vector (Vector Shedding), medido en muestras de orina, saliva y heces después de la infusión [Marco de tiempo: Día 1 hasta la semana 104].
Nivel de títulos de anticuerpos contra el virus adenoasociado recombinante serotipo rh74 (rAAVrh74) [Marco de tiempo: Día 2 hasta la semana 156].
Número de participantes con eventos adversos emergentes del tratamiento (TEAE), eventos adversos graves (SAE) y eventos adversos de especial interés (AESI) [Marco de tiempo: Inicio hasta la semana 156].
Cohorte 8: Número de participantes con infecciones, edema, complicaciones en la cicatrización de heridas, hiperlipidemia, angioedema y enfermedad pulmonar intestinal/neumonitis no infecciosa [Marco de tiempo: Inicio hasta la semana 72].
Cambio desde el inicio en la cantidad de expresión proteica de distrofina de delandistrogene moxeparvovec a la semana 12, medido por intensidad de fibra mediante inmunofluorescencia (IF) [Marco de tiempo: Inicio, Semana 12].
Cambio desde el inicio en la cantidad de expresión proteica de distrofina de delandistrogene moxeparvovec a la semana 12, medido por porcentaje de fibras positivas para distrofina (PDPF) mediante IF [Marco de tiempo: Inicio, Semana 12].
Cantidad de expresión proteica de distrofina de delandistrogene moxeparvovec a la semana 12, medido por intensidad de fibra mediante IF [Marco de tiempo: Semana 12].
Cantidad de expresión proteica de distrofina de delandistrogene moxeparvovec a la semana 12, medido por PDPF mediante IF [Marco de tiempo: Semana 12].
Cohorte 8: Número de participantes con eventos adversos hepáticos, biomarcadores hepáticos y evaluaciones de laboratorio indicativas de lesión hepatocelular aguda o disfunción hepática aguda [Marco de tiempo: Inicio hasta la semana 72].
Cohorte 8: Número de participantes con ALI grave [Marco de tiempo: Inicio hasta la semana 72].
Cohorte 8: Duración de los esteroides administrados [Marco de tiempo: Inicio hasta la semana 72].
Aquí lo más importante es el cohorte 8, donde se medirá la respuesta
inmune y la toxicidad hepática del tratamiento por las muertes del
2025 de dos adolescentes tratados y un niño.
NCT05096221 – EMBARK, trial 301
Un fase 3, RCT, doble ciego.
AAV gene therapy for Duchenne muscular dystrophy: the EMBARK phase 3 randomized trial
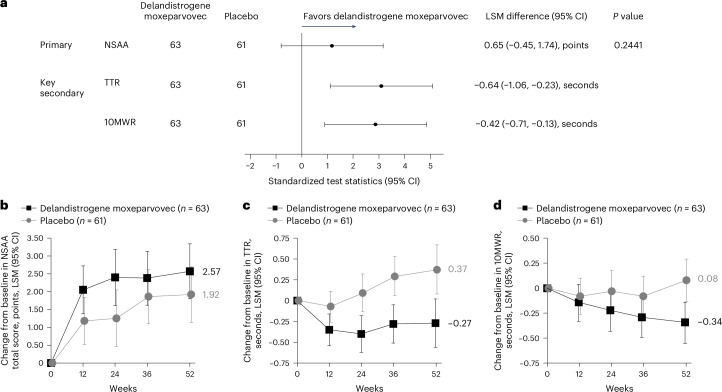
The primary outcome was change in NSAA total score from baseline to week 52 (Part 1). The NSAA is a categorical assessment of motor function in ambulatory patients with DMD, consisting of 17 items scored with a 0, 1 or 2 based on the patient’s ability to complete the task. At week 52 in the overall population, the least squares mean (LSM) change (95% confidence interval (CI)) from baseline in NSAA total score was 2.57 (1.80, 3.34) versus 1.92 (1.14, 2.70) points with delandistrogene moxeparvovec and placebo, respectively. The between-group difference was not statistically significant (0.65 (s.e. = 0.55) points; 95% CI, −0.45, 1.74; P = 0.2441; Fig. 2a,b). Results were consistent across pre-specified age subgroups and baseline NSAA total score subgroups (Supplementary Table 2).
Secondary outcomes
As defined per protocol, key secondary functional endpoints were Time to Rise (TTR) from the floor and 10-meter Walk/Run (10MWR) at week 52. The LSM change (95% CI) from baseline to week 52 on the TTR was −0.27 s (−0.56, 0.02) for delandistrogene moxeparvovec versus 0.37 s (0.08, 0.67) for placebo, with a between-group difference of −0.64 s (95% CI, −1.06, −0.23). Similarly, the LSM change (95% CI) from baseline to week 52 on the 10MWR was −0.34 s (−0.55, −0.14) for delandistrogene moxeparvovec versus 0.08 s (−0.13, 0.29) for placebo, with a between-group difference of –0.42 s (95% CI, −0.71, −0.13) (Fig. 2a,c,d). Subgroup analysis data are provided in Supplementary Table 2.
Other secondary functional endpoints assessed were stride velocity 95th centile (SV95C), 100-meter Walk/Run (100MWR) and time to ascend 4 steps. The LSM change (95% CI) from baseline to week 52 on SV95C was 0.06 meters per second (0.00, 0.13) for delandistrogene moxeparvovec versus –0.03 meters per second (−0.09, 0.03) for placebo, with a between-group difference of 0.10 meters per second (95% CI, 0.00, 0.19). The LSM change (95% CI) from baseline to week 52 on the 100MWR was −6.57 s (−10.05, −3.09) for delandistrogene moxeparvovec versus −3.28 s (−6.86, 0.29) for placebo, with a between-group difference of −3.29 s (95% CI, −8.28, 1.70). Analysis of time to ascend 4 steps showed LSM change (95% CI) from baseline to week 52 of −0.44 s (−0.69, −0.20) for delandistrogene moxeparvovec versus −0.08 s (−0.33, 0.17) for placebo, with a between-group difference of −0.36 s (95% CI, −0.71, −0.01) (Fig. 3). Subgroup analyses by age and baseline NSAA total scores are presented in Supplementary Table 2.
Two-Year Outcomes Following Delandistrogene Moxeparvovec Treatment in Ambulatory Patients with Duchenne Muscular Dystrophy: Phase 3 EMBARK Trial
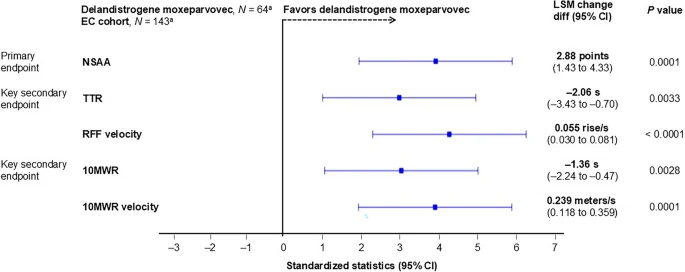
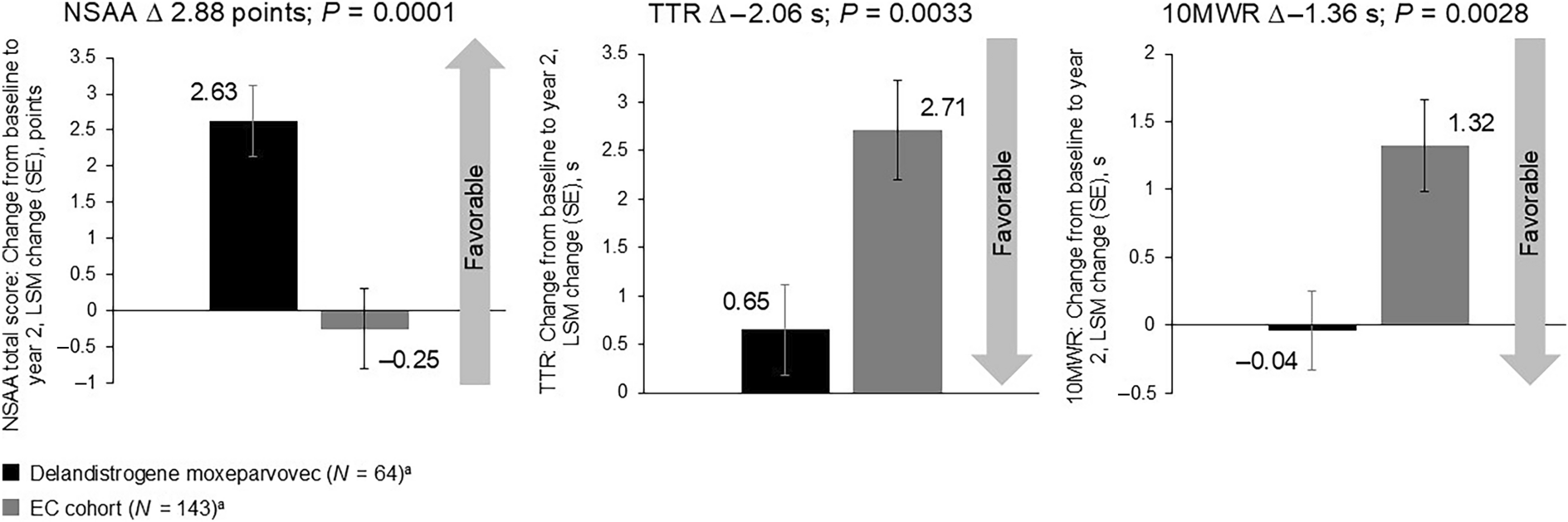
At 2 years, patients treated with delandistrogene moxeparvovec (n = 63; one patient treated in EMBARK part 1 did not have 2-year follow-up data) demonstrated statistically significant functional benefit versus the EC cohort (Figs. 1 and 2) in key functional outcomes of change from baseline for NSAA total score (2.63 versus − 0.25 points; LSM [95% CI] 2.88 [1.43–4.33]; P = 0.0001), TTR (0.65 versus 2.71 s; − 2.06 [− 3.43 to − 0.70]; P = 0.0033), and 10MWR (− 0.04 versus 1.32 s; − 1.36 [− 2.24 to − 0.47]; P = 0.0028); all P values are nominal and have not been adjusted for multiple comparisons. Change from baseline in RFF velocity (0.003 versus − 0.053 rise/s; 0.055 [0.03–0.08]; P < 0.0001) and 10MWR velocity (0.124 versus − 0.115 m/s; 0.239 [0.12–0.36]; P = 0.0001) also demonstrated statistically significant functional benefit versus the EC cohort (see Fig. S1 in the Supplementary Material).
Forest plot of functional outcomes 2 years post-delandistrogene moxeparvovec infusion compared with the EC cohort. aAll 64 patients treated with delandistrogene moxeparvovec and all 143 patients in the EC cohort were included in the analyses; MMRM methods account for missing data in these analyses. One patient treated with delandistrogene moxeparvovec in part 1 did not have 2-year follow-up data (the part 2 week 52 NSAA was deemed invalid because of behavioral reasons). In the EC cohort, 29, 28, and 30 patients had missing year 2 data for NSAA, TTR, and 10MWR assessments, respectively. Negative values for timed function tests (TTR, 10MWR) show an improvement in the time taken to achieve these endpoints. All P values are nominal and not adjusted for multiple comparisons. LSMs (of change from baseline) and CIs were standardized by dividing by the SE. LSM differences are on the original scale (without SE adjustment). Signs of timed function tests were reversed in the forest plot to align favorable directions among endpoints. Numerical results of LSM difference kept the original signs. 10MWR 10-m Walk/Run, CI confidence interval, EC external control, RFF rise from floor, LSM least-squares mean, MMRM mixed effects models for repeated measures, NSAA North Star Ambulatory Assessment, s seconds, SE standard error, TTR Time to Rise
Baseline to year 2 LSM changes in functional outcomes post-delandistrogene moxeparvovec infusion compared with the EC cohort. aAll 64 patients treated with delandistrogene moxeparvovec and all 143 patients in the EC cohort were included in the analyses; MMRM methods account for missing data in these analyses. One patient treated with delandistrogene moxeparvovec in part 1 did not have 2-year follow-up data (the part 2 week 52 NSAA was deemed invalid because of behavioral reasons). In the EC cohort, 29, 28, and 30 patients had missing year 2 data for NSAA, TTR, and 10MWR assessments, respectively. Negative values for timed function tests (TTR, 10MWR) show an improvement in the time taken to achieve these endpoints. All P values are nominal and not adjusted for multiple comparisons. 10MWR 10-m Walk/Run, EC external control, LSM least-squares mean, MMRM mixed effects models for repeated measures, NSAA North Star Ambulatory Assessment, s seconds, SE standard error, TTR Time to Rise
Biological Outcomes
Analyses of muscle biopsies performed in patients treated at a subset of sites (preselected on the basis of experience in performing muscle biopsies) at week 12 (n = 17) and week 64 (n = 16; one patient was transferred to a non-biopsy site in part 2 and a week 64 biopsy was not collected) demonstrated sustained micro-dystrophin expression and sarcolemmal localization. Mean (standard deviation) western blot (% control) was 34.29% (41.04%) at week 12 and 45.68% (39.75%) at week 64. The percentage of dystrophin-positive fibers was 28.13% (26.10%) at week 12 and 38.60% (26.93%) at week 64.
Results
At 2 years, EMBARK patients showed statistically significant benefit versus the EC cohort in functional outcomes prognostic for delaying loss of ambulation (NSAA, Time to Rise, 10-m Walk/Run), demonstrating sustained stabilization or slowing of disease progression. Delandistrogene moxeparvovec micro-dystrophin expression and sarcolemmal localization were maintained over 64 weeks. No new safety signals were observed between week 52 and week 104. Between baseline and week 104, there were no treatment-related deaths, study discontinuations due to adverse events, or clinically significant complement-mediated adverse events.
Conclusions
At 2 years, stabilization or slowing of DMD disease progression was observed in ambulatory male patients with DMD aged 4 to < 8 years receiving delandistrogene moxeparvovec versus a matched EC cohort. Safety was consistent with EMBARK 1-year data and manageable with appropriate monitoring.
--(BUSINESS WIRE)-- (NASDAQ:SRPT), the leader in precision genetic medicine for rare diseases, today announced positive topline three-year functional results from Part 1-treated patients in EMBARK (Study SRP-9001-301), the global, randomized placebo-controlled Phase 3 study evaluating ELEVIDYS (delandistrogene moxeparvovec-rokl) in ambulatory individuals with Duchenne muscular dystrophy who were aged four to seven at time of treatment and at time of last assessment were on average over nine years of age.
Three years after treatment, patients who received ELEVIDYS in Part 1 of EMBARK demonstrated statistically significant, clinically meaningful and durable efficacy across all key motor function measures, North Star Ambulatory Assessment (NSAA), Time to Rise (TTR) and 10-meter walk/run (10MWR), when compared to a pre-specified propensity-weighted untreated external control group (EC)*. The mean NSAA score remained above baseline at Year 3 for the ELEVIDYS-treated group (n=52) while the EC group (n=73) continued to show the expected age-related decline below their baseline score. The ELEVIDYS group showed a 73% slowing of disease progression as measured by TTR and 70% slowing of disease progression as measured by 10MWR when compared to the EC group.**
Patients treated with ELEVIDYS in Part 1 maintained significantly higher levels of motor function three years after treatment compared to the EC group. Topline efficacy results are summarized in the table below:
“ELEVIDYS is the first gene therapy for Duchenne to show a dramatic shift in disease trajectory out to three years consistent with earlier long-term data extending up to five years. This is long-term data in a robust, controlled clinical dataset that demonstrates the power of a disease-modifying therapy targeting the underlying cause of Duchenne,” said Louise Rodino-Klapac, Ph.D., president of research & development and technical operations, Sarepta. “At an age when functional decline is typically accelerating, ELEVIDYS-treated patients showed a 70 percent or greater reduction in the rate of decline on key functional measures such as time to rise and the 10-meter walk/run. These statistically significant benefits not only persist but continue to strengthen over time, creating a sustained and growing separation from the expected disease trajectory.”
“As a pediatric neurologist, I spend time with families who are doing everything they can to help their children stay strong in the face of Duchenne,” said Crystal Proud, M.D., chief of Neurology and director of Neuromuscular Medicine at Children’s Hospital of The King’s Daughters, and an investigator in the EMBARK study. “The EMBARK results give us a clearer picture of how treatment with ELEVIDYS can make a meaningful difference over time, and they reflect what I see in clinical practice – helping boys perform everyday movements, such as standing, walking and running with greater strength and speed than what we expect as Duchenne progresses without a disease-modifying treatment.”
No new treatment-related safety signals were observed, reinforcing the consistent and manageable safety profile seen in ambulatory patients treated with ELEVIDYS to date. Analysis of the three-year data is ongoing and includes functional results from crossover-treated patients two years after treatment. The Company plans to share results at upcoming medical meetings and in publication. Two-year EMBARK results were published in Neurology & Therapy this month.
NCT05881408 – ENVISION
Primary Completion Date (Estimated):
2027-05-31 (Final data collection date for primary outcome measure)
Study Completion Date (Estimated): 2028-06-30
The study will evaluate the safety and efficacy of delandistrogene moxeparvovec gene transfer therapy in non-ambulatory and ambulatory males with DMD. This is a randomized, double-blind, placebo-controlled 2-part study. Participants will be in the study for approximately 128 weeks. All participants will have the opportunity to receive intravenous (IV) delandistrogene moxeparvovec in either Part 1 or Part 2.
Inclusion Criteria:
Definitive diagnosis of DMD based on documented clinical findings and prior genetic testing.
Cohort 1 only: Non-ambulatory per protocol-specified criteria.
Cohort 2 only: Ambulatory per protocol-specified criteria and ≥8 to <18 years of age at the time of Screening.
Ability to cooperate with motor assessment testing.
Stable daily dose of oral corticosteroids for at least 12 weeks prior to Screening, and the dose is expected to remain constant throughout the study (except for modifications to accommodate changes in weight).
Recombinant Adeno-Associated Virus Serotype rh74 (rAAVrh74) antibody titers are not elevated as per protocol-specified requirements.
A pathogenic frameshift mutation or premature stop codon in the DMD gene, except for any deletion mutations in exon 8 and/or 9.
Exclusion Criteria:
Exposure to gene therapy, investigational medication, or any treatment designed to increase dystrophin expression within protocol specified time limits.
Abnormality in protocol-specified diagnostic evaluations or laboratory tests.
Presence of any other clinically significant illness, medical condition, or requirement for chronic drug treatment that in the opinion of the Investigator creates unnecessary risk for gene transfer.
Ages (Child, Adult, Older Adult)
Primary (Current) *ICMJE (Submitted: 2023-05-19)
Part 1: Change From Baseline in the Total Score of Performance of Upper Limb (PUL) (Version 2.0) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Percent Predicted Forced Vital Capacity (FVC) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Percent Predicted Peak Expiratory Flow (PEF) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Quantity of Delandistrogene Moxeparvovec Dystrophin Expression at Week 12 as Measured by Western Blot [Time Frame: Week 12]
Part 1: Change From Baseline in Patient-Reported Outcomes Measurement Information (PROMIS) Score in Upper Extremity Function to Week 72 [Time Frame: Baseline, Week 72]
Number of Participants with a Treatment Emergent Adverse Event (TEAE), Adverse Event of Special Interest (AESI), and Serious Adverse Event (SAE) [Time Frame: Baseline up to Week 124]
Part 1 (For Cohort 2 Only): Change From Baseline in the North Star Ambulatory Assessment (NSAA) Total Score at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Global Circumferential Strain as Measured by Cardiac MRI at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in the Middle Domain Score of PUL (Version 2.0) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Percent Predicted Forced Vital Capacity (FVC) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Percent Predicted Peak Expiratory Flow (PEF) at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Quantity of SRP-9001 Protein Expression at Week 12 as Measured by Western Blot [Time Frame: Week 12]
Part 1: Change From Baseline in Patient-Reported Outcomes Measurement Information (PROMIS) Score in Upper Extremity Function to Week 72 [Time Frame: Baseline, Week 72]
Number of Participants with a Treatment Emergent Adverse Event (TEAE), Adverse Event of Special Interest (AESI), and Serious Adverse Event (SAE) [Time Frame: Baseline up to Week 124]
Part 1 (For Cohort 2 Only): Change From Baseline in the North Star Ambulatory Assessment (NSAA) Total Score at Week 72 [Time Frame: Baseline, Week 72]
Part 1: Change From Baseline in Global Circumferential Strain as Measured by Cardiac MRI at Week 72 [Time Frame: Baseline, Week 72]
Con esto podemos deducir que ENVISION busca medir los resultados y
qué tan tolerable es en edades mayores a 7 años, junto a su evolución.
Clave para la comercialización del tratamiento.
6. Alcance del Tratamiento
Con el libro sabemos que la enfermedad es muy individual (variable en
su evolución), que la pérdida de movilidad llevando a necesitar silla de
ruedas es predictivo de problemas cardíacos y pulmonares, que los
resultados de NSAA crece hasta los 6-7 años (la edad de placebo de
estudios de Elevidys) y después disminuye.
Sobre los fallecimientos de pacientes
Dos casos fatales fueron insuficiencia hepática aguda, dos adolescentes
(no ambulatorios) fallecieron poco después de recibir el tratamiento.
Esto es conocido en tratamientos con vectores virales en terapias
genéticas. Uno de ellos había sufrido de una infección por citomegalovirus
que podía haber contribuido con la inflamación de hígado antes o durante
el tratamiento.
Un niño de 8 años murió por influenza, que pudo ser agravado por el
tratamiento de corticoides para ser tratado con Elevidys. Sufrió de una
falla multiorgánica, se determinó que no hubo vinculo con Elevidys.
En los casos de insuficiencia aguda ellos tenían deleciones completas
en los exones 8 y 9, que otros pacientes también presentan, con lo que
parece ser que el problema es una combinación de factores en los pacientes
que los hace propensos a respuestas inmunes a la micro-distrofina que
termina atacando a los músculos y corazón. Un factor posible es el de
poseer un HLA con afinidad química por proteína producida por los
exones 8 y 9, provocando una respuesta inmune agresiva que no es
posible controlar con corticoides. También, algunos pacientes ya tienen
células T contra secuencias similares, causando la respuesta inmune.
Es por esto que se está siendo prudente eliminando a pacientes con
deleciones completas de los exones 8 y 9 y el porqué el cohorte 8 de
ENVISION es clave para poder controlar los efectos auto-inmunes y
saber que factores en combinación son los que realmente provocan
respuestas inmunes agresiva contra la micro-distrofina en los tejidos.
Aún así, estas muertes, aunque lamentables, suelen ser comunes en
tratamientos por AAV de alteración genética y la reacción del mercado
ante estos acontecimientos parecen muy exageradas tomando en cuenta
el resultado del tratamiento y los puntuales casos con características muy
distintivas. Quizá la desconfianza de aceptación por parte de la FDA y el
estar al borde de errar fuertemente y ser la compañía en vanguardia puede
explicar estos miedos, además de un estado pesimista e incluso obsesivo en
contra de la empresa y el Elevidys. Esto se expresa incluso a desconfianza
en Doug (CEO) y sus capacidades, un tanto exagerado en mi opinión, pero
no es que tenga décadas de experiencia así que quizá tengan razón.
Pero me voy a limitar a pensar en lo que tengo seguridad. El tratamiento
funciona después de 2-3 años y mientras más joven mejor, no es posible
tratar a pacientes con deleciones de los exones 8 y 9, hasta ahora no es
aconsejable tratar a pacientes no ambulatorios. Eso es lo que tenemos para
hacer el análisis.
La proporción de mutaciones de DMD es de 1:5000, la de deleciones
completas de exones 8 y 9 es de entre el 3 y 5% de los casos de DMD,
por tanto, el ratio es de 1:5250 para pacientes tratables, además de que
debe ser corregido por demografía de 4-7 años para cada zona a tratar y
anticuerpos contra AAVrh74 (~15%). Ratio de ~1:6040.
Como los ensayos clínicos suelen ser presentes en EEUU, Europa, Japón
y Brasil serán usados como poblaciones objetivo.
EE.UU. será una rápida adopción por tener una infraestructura ya instalada
en centros especializados y la presión de Doug para estar listos como
mencionó en la presentación del 26 de enero. Voy a suponer una adopción
del 90% de los niños a tercer año. Población de 4-7 años: ~14.4 millones,
con DMD aceptables: 2380.
Europa es mucho más conservador y lo demostraron con los términos de
estudios clínicos en varios países. El primer año habrá cero niños
tratados y a tercer año 30%. Población de 4-7 años: ~18.2 millones,
con DMD aceptables: 3000.
Brasil una adopción lenta por alto costo, primer periodo un 5-10% con
crecimiento moderado por judicializaciones. Población de 4-7 años:
~10.5 millones, con DMD aceptables: 1700.
Japón tendrá una adopción constante pero conservadora de 40 niños el
primer periodo y crecerá constantemente. Población de 4-7 años:
~2.9 millones, con DMD aceptables: 480.
7. Modelo de Valoración
Ingresos
La producción y distribución de Elevidys tiene varias partes y sigue la
estrategia de delegar ex-US a un tercero. Hay regalías de propiedad
intelectual que debe pagar a Nationwide Children’s Hospital (NCH)
por usar AAVrh74, al rededor de 1-3% de las ventas netas globales.
Alianza Sarepta-Roche
Roche abona pagos en cash y Equity para tener derechos sobre ex-US
junto a reparto de Costos 50/50. Las regalías a pagar a Sarepta es del
13-17% sobre ventas totales.
Producción
Catalent, Inc. se encarga de ser el fabricante primario de Elevidys. El
2025 no pasó protocolos de higienización de la línea de Elevidys, por
buscar mantener costos bajos en una industria muy volatil. Esto
llevó a despedir 350 empleados en Maryland. Se retrasó la producción
de lotes de Elevidys a 2027.
Thermo Fisher Scientific suministra plásmidos para la producción de
del vector.
Arrowhead Pharmaceuticals
Alianza para distribución global de tratamientos de siRNA. The clinical-stage programs covered under the agreement include:
ARO-DUX4: designed to reduce the production of human double homeobox 4 (DUX4) protein in skeletal muscle; currently in a Phase 1/2 clinical study for the treatment of facioscapulohumeral muscular dystrophy (FSHD)
ARO-DM1: designed to target and suppress myotonic dystrophy protein kinase (DMPK) in skeletal muscle; Phase 1/2 clinical study for myotonic dystrophy type 1 (DM1)
ARO-MMP7: designed to reduce expression of matrix metalloproteinase 7 (MMP7) in pulmonary epithelial cells; Phase 1/2 clinical study for idiopathic pulmonary fibrosis (IPF)
ARO-ATXN2: designed to target the ataxin-2 protein (ATXN2) in the CNS; expected to begin Phase 1/2 clinical study for spinocerebellar ataxia 2 (SCA2) by the end of 2024
Precios
EE.UU.
El precio de dosis es de $3.2 millones, sin embargo por regulación será de
alrededor de $2.5 millones.
Europa
No hay aprobación pero, en 2025 en rusia era de alrededor de 2.6 a 2.2
millones de euros. Así que poner un precio de 2.5 millones de euros no
parece muy lejos de la realidad. Si Roche es 13% en ingreso de 0.352m
y si es 17% es de 0.425m.
Japón
Cerca de 500M yen, ~$3.6MM, con rango de Roche: $0.468MM y
$0.612MM.
Brasil
Cerca de17MM reales, con un descuento del 20% por demandas, queda
en 13.6MM. Para Sarepta, en dólares, entre ~0.338MM y ~0.442MM.
A todas estas ventas el descuento de regalías de 1-3% a NCH
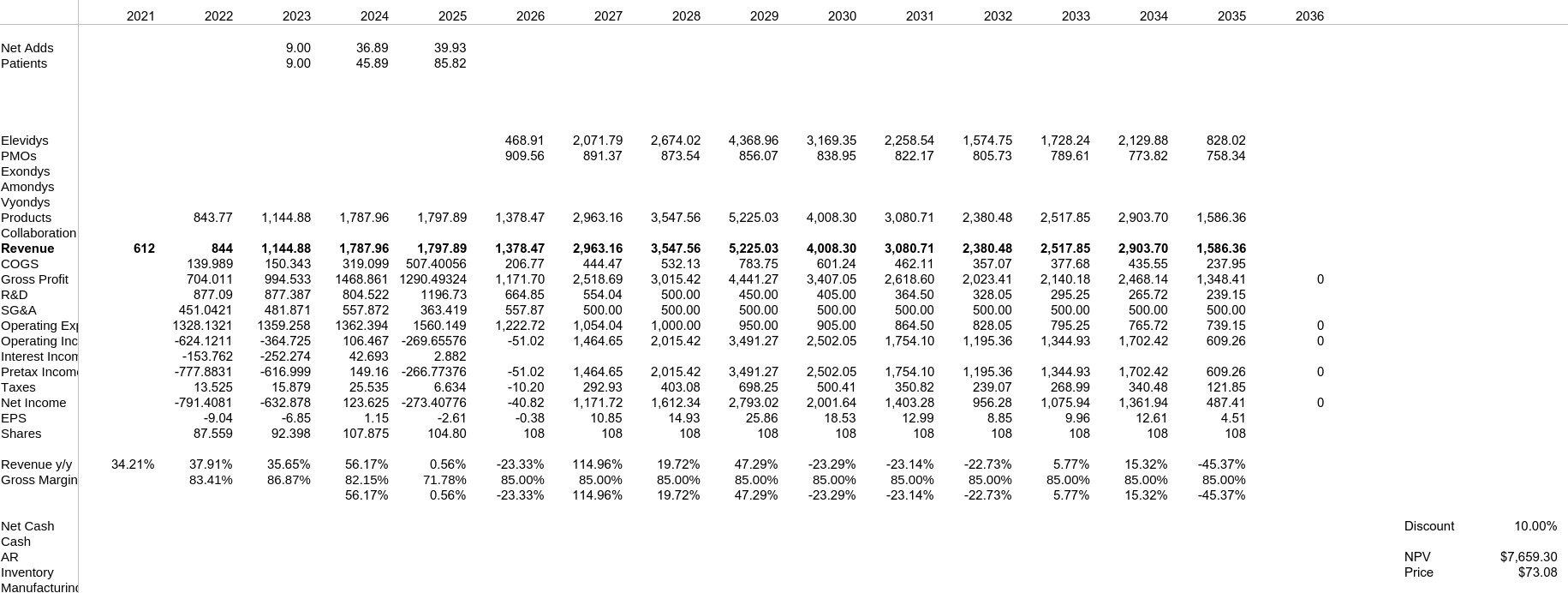
Sheets
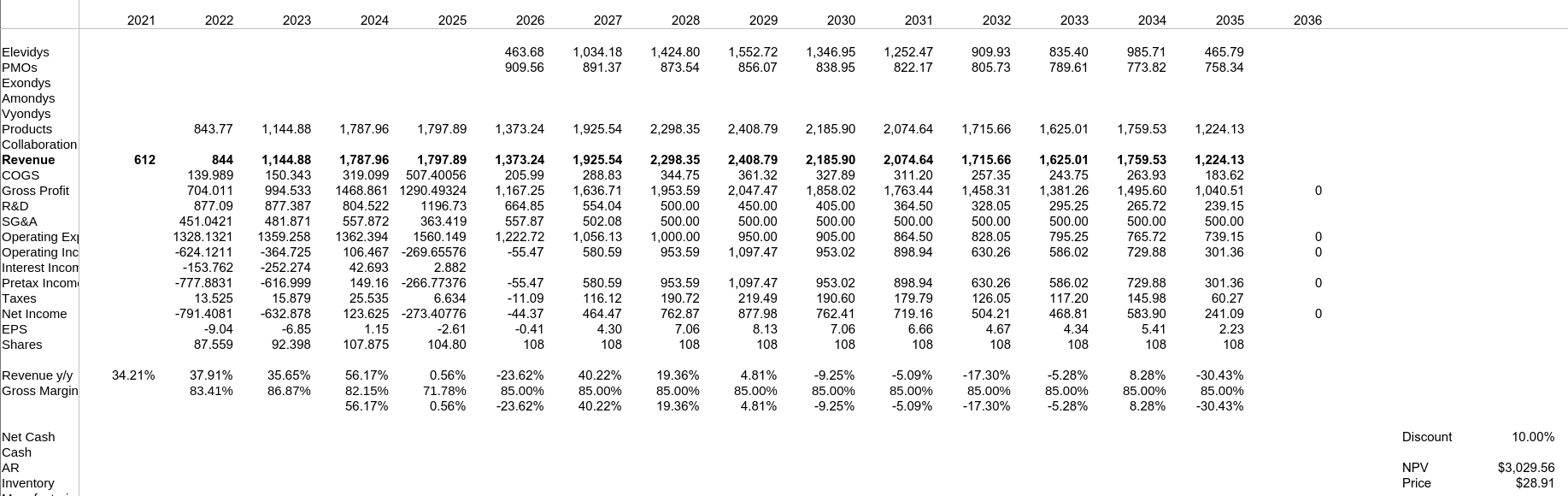
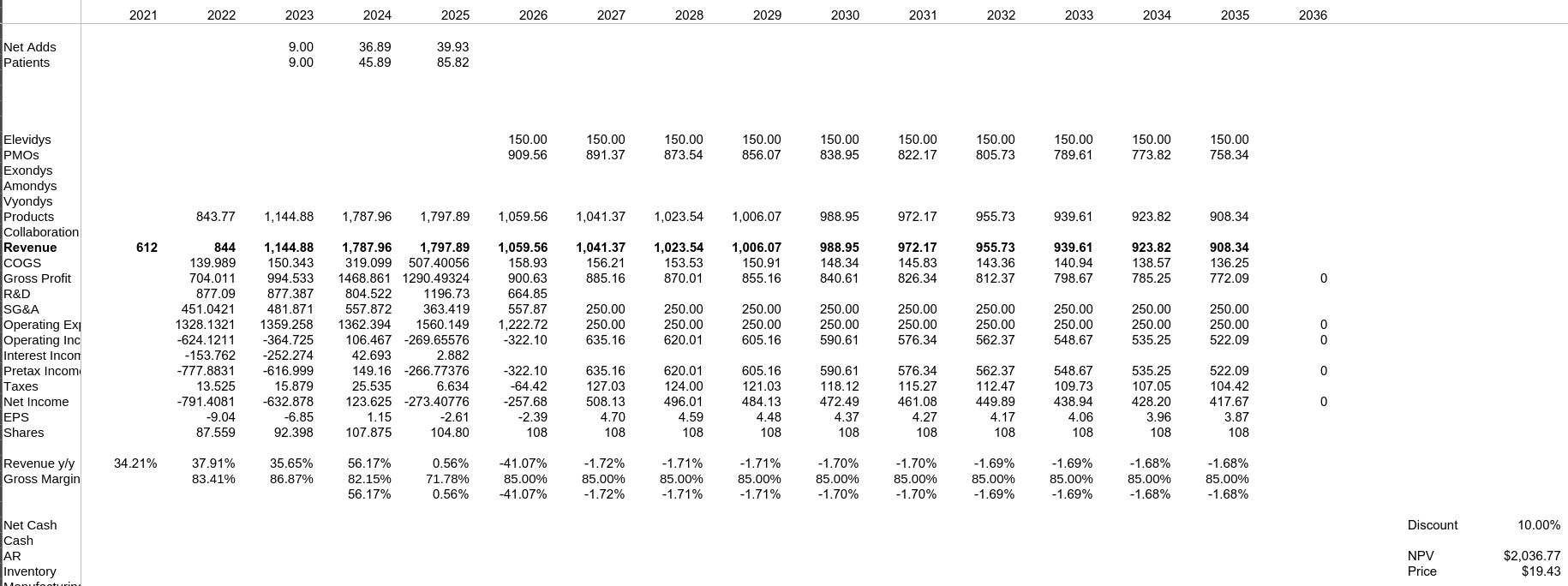
Modelo Base
Modelo Black Label
Modelo Éxito - No Black Label y Disponible en No Ambulatorios
8. Conclusión
Elevidys es un tratamiento de vanguardia ante un enfermedad genética
que es persistente en la población y que, mediante un mecanismo de AAV,
es capaz de producir suficiente micro-distrofina para encontrar
diferencias notorias a los 3 años de la inyección de la dosis. Por ahora,
no es aconsejable el uso en personas mayores a 7 años con deleciones en
los exones 8 y 9 del gen de distrofina, sin embargo el problema es una
combinación de esto y variabilidad genética individual, es por eso que
ENDEAVOR es clave en la superación de este problema y poder suministrar
el tratamiento a personas mayores a 7 años con cuidados constantes con
los que medir toxicidad de respuesta auto-inmune con el cohorte 8.
El tratamiento funciona y la toxicidad es muy individual, está la posibilidad
de que los resultados de mediados de año muestren incapacidad de tratar
a niños mayores, por lo que el precio no será mayor de $30, en cambio,
si resulta que es manejable y tolerable se disparará a $70, por lo bajo.
Tomar decisiones siendo consciente de los riesgos y que el tamaño de
las posiciones que uno toma son la mejor forma de manejar riesgo.